Safety Assessments
Safety assessments included collection of treatment-emergent adverse events (TEAEs) throughout the study, and physical examination of the face, head, and neck at baseline, day 2, week 2, month 1, and month 6 after treatment.
Statistics
Statistical calculations were done using SAS® version 9.4. Efficacy analyses were based on the intent-to-treat (ITT) population, defined as all subjects who were randomized, or on the per-protocol (PP) population, defined as an ITT subject with no protocol deviations with substantial impact on primary efficacy outcome. The safety population consisted of all subjects administered study drug.
Primary endpoint: the composite 2-grade responder rate at maximum frown at month 1 for aboBoNT-A and placebo were compared using Cochran-Mantel-Haenszel test stratified by center at a 5% significance level (2-sided). Pooling of centers was done based on geographical location, until the pooled center had at least 16 subjects, and at least one responder and one non-responder for the primary endpoint. Missing data were handled by multiple imputation. The secondary and exploratory ILA and SSA responder rates were compared using Cochran- Mantel-Haenszel test stratified by pooled center.
For analysis of duration of effect and time to onset of treatment response, Kaplan-Meier estimates of the median event times were used.
For the two FACE-Q scales containing multiple items, the subjects’ scores for the individual items were converted to a single Rasch-transformed total score from 0 to 100 for each scale as per the FACE-Q manual. Higher total scores indicated greater psychological function or that subjects were less bothered by their GL appearance. No statistical comparisons were performed for FACE-Q data.
Safety assessments included collection of treatment-emergent adverse events (TEAEs) throughout the study, and physical examination of the face, head, and neck at baseline, day 2, week 2, month 1, and month 6 after treatment.
Statistics
Statistical calculations were done using SAS® version 9.4. Efficacy analyses were based on the intent-to-treat (ITT) population, defined as all subjects who were randomized, or on the per-protocol (PP) population, defined as an ITT subject with no protocol deviations with substantial impact on primary efficacy outcome. The safety population consisted of all subjects administered study drug.
Primary endpoint: the composite 2-grade responder rate at maximum frown at month 1 for aboBoNT-A and placebo were compared using Cochran-Mantel-Haenszel test stratified by center at a 5% significance level (2-sided). Pooling of centers was done based on geographical location, until the pooled center had at least 16 subjects, and at least one responder and one non-responder for the primary endpoint. Missing data were handled by multiple imputation. The secondary and exploratory ILA and SSA responder rates were compared using Cochran- Mantel-Haenszel test stratified by pooled center.
For analysis of duration of effect and time to onset of treatment response, Kaplan-Meier estimates of the median event times were used.
For the two FACE-Q scales containing multiple items, the subjects’ scores for the individual items were converted to a single Rasch-transformed total score from 0 to 100 for each scale as per the FACE-Q manual. Higher total scores indicated greater psychological function or that subjects were less bothered by their GL appearance. No statistical comparisons were performed for FACE-Q data.
RESULTS
Subject Disposition and Demographics
In total, 301 subjects were randomized, comprising the ITT population, 300 were treated, comprising the safety population, and 287 subjects (95%) completed the study. Most noncompleters were lost to follow-up. No subjects discontinued due to adverse events. One subject in the aboBoNT-A group was randomized in violation of the age criteria (aged 65) and was therefore withdrawn before receiving
treatment.
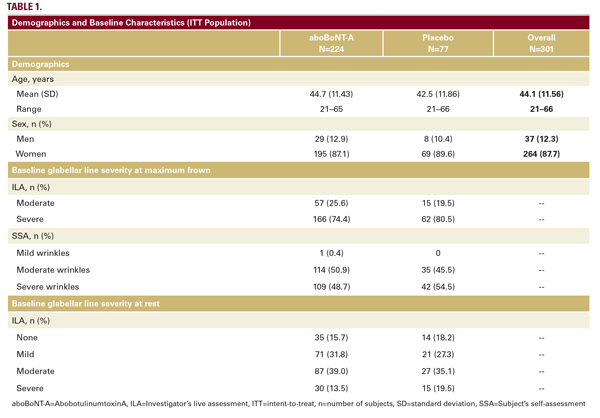
In total, 301 subjects were randomized, comprising the ITT population, 300 were treated, comprising the safety population, and 287 subjects (95%) completed the study. Most noncompleters were lost to follow-up. No subjects discontinued due to adverse events. One subject in the aboBoNT-A group was randomized in violation of the age criteria (aged 65) and was therefore withdrawn before receiving
treatment.

