INTRODUCTION
Granulomatous dermatoses (GDs) are a group of diseases defined by organized granuloma formation within the dermis or hypodermis, often in response to infections, inflammation, neoplasia, or metabolic disorders.1 Granulomas are thought to arise as a delayed hypersensitivity reaction to persistent antigens that cannot be eliminated by phagocytosis.1 They consist of aggregates of activated macrophages that differentiate into epithelioid histiocytes, sometimes accompanied by multinucleated giant cells and dendritic cells, and a surrounding lymphocytic border.1 Granulomatous inflammation is primarily driven by a T-helper (Th)1 response, via interferon (IFN)γ, tumor necrosis factor (TNF)-α, interleukin (IL)-2, and in some contexts, Th17 involvement.1 While the immunologic basis of granuloma formation is shared across GDs, their clinical presentations, histopathologic nuances, and therapeutic responses differ. This review focuses on non-infectious GDs, including granuloma annulare (GA), sarcoidosis, and interstitial granulomatous dermatitis (IGD), highlighting their clinicopathologic overlap, distinguishing features, and management approaches.
Granuloma Annulare
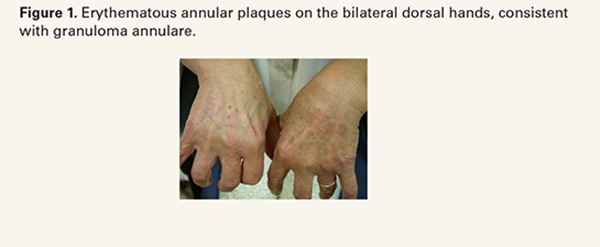
GA is the most common non-infectious GD, with an estimated prevalence of 0.04% in the United States (U.S.).2,3 Clinically, GA presents as asymptomatic, annular, erythematous papules or plaques, most commonly affecting the dorsal and lateral aspects of the hands and feet (Figure 1).3 Most cases of GA are localized; however, other subtypes include generalized, perforating, patch, and subcutaneous forms.3 The exact etiology of GA remains incompletely understood, but has classically been considered a Th1-mediated delayed-type hypersensitivity reaction, in which IFN- γ-activated macrophages release matrix metalloproteinases leading to collagen degradation.3,4 More recent data also suggests potential Th2 involvement.3,4 GA has been associated with diabetes mellitus, hyperlipidemia, autoimmune disease (rheumatoid arthritis and systemic lupus erythematosus (SLE)), thyroid disease, vaccinations, infections, and iatrogenic triggers.3,4 Population-level data support baseline metabolic and thyroid screening, particularly in generalized
or atypical presentations, while routine malignancy workup beyond age-appropriate guidelines is not indicated unless disease is recalcitrant or systemic.2 Diagnosis is primarily clinical but may be confirmed histologically, with biopsy demonstrating focal collagen degeneration, mucin deposition, and an interstitial histiocytic infiltrate.3
Although GA is benign and often self-limiting, recurrence is common and therapeutic response is often variable.3 First-line management includes intralesional (IL) and topical corticosteroids (TCS).2-4 Other common treatments include tetracyclines, hydroxychloroquine, and phototherapy.2 Janus kinase (JAK) inhibitors have been reported as emerging therapies, with JAK1/3 inhibitor oral tofacitinib 5 mg twice daily demonstrating clinical and histologic remission in recalcitrant GA.5 Delgocitinib 0.5% ointment, a pan-JAK inhibitor, has demonstrated preliminary efficacy after two months of twice-daily application, and ruxolitinib 1.5% cream, a selective JAK1/2 inhibitor, has been reported to achieve complete clearance of GA after three months of treatment.6,7 Additionally, a 2026 retrospective cohort trial found that the Goeckerman protocol (coal tar, phototherapy, and TCS) significantly outperformed TCS (56.3%), combination TCS + phototherapy (69.6%), and phototherapy alone in treating GA (16.7%).8 On multivariate analysis, the Goeckerman protocol was the strongest predictor of treatment response (Odds Ratio (OR) 32.52; P < 0.001).8 Other therapies, including apremilast, TNF-α inhibitors, methotrexate, isotretinoin, vitamin E, topical imiquimod, dapsone, and systemic retinoids, have been reported in small case series and case reports, but robust evidence supporting consistent efficacy remains limited.3,4
Sarcoidosis
Sarcoidosis is a multisystem inflammatory GD characterized by the formation of non-caseating granulomas that can involve nearly every organ system, most commonly the lungs and skin.9,10 In the U.S., sarcoidosis disproportionately affects Black individuals, with a consistent female predominance across populations.9 Although the pathogenesis of sarcoidosis is not fully understood, it is believed to arise from a combination of genetic susceptibility, immune dysregulation, and environmental exposure.9
Cutaneous involvement occurs in approximately 20-30% of patients and may represent the initial manifestation of systemic disease.9,10 Specific cutaneous lesions are a result of granulomatous inflammation and often present as papules, plaques, lupus pernio, subcutaneous nodules, or scar sarcoidosis (Figure 2).9,10 Given its

