Lichen Myxedematosus: Case Report and Review of Literature
March 2020 | Volume 19 | Issue 3 | Case Reports | 320 | Copyright © March 2020
Published online February 5, 2020
Amaris Geisler BS,a Mojgan Hosseinipour DO,b Nikki S. Vyas MD,c Robert Phelps MD,d Charles Gropper MD,e Cindy Hoffman DOf
aMedical Student, CUNY School of Medicine, New York, NY bResident Physician, St. Barnabas Hospital, Department of Dermatology, Bronx, NY cResident Physician, Department of Pathology, Mount Sinai Medical Center, New York, NY dDirector of Dermatopathology, Mount Sinai Medical Center, New York, NY eChief of Dermatology, St. Barnabas Hospital, Bronx, NY fProgram Director, St. Barnabas Hospital, Department of Dermatology, Bronx, NY
Abstract
Lichen myxedematosus (LM) is an idiopathic cutaneous mucinosis, commonly described as localized scleromyxedema. In contrast to scleromyxedema, there is typically no systemic involvement. Treatment options are limited and spontaneous resolution has been reported.
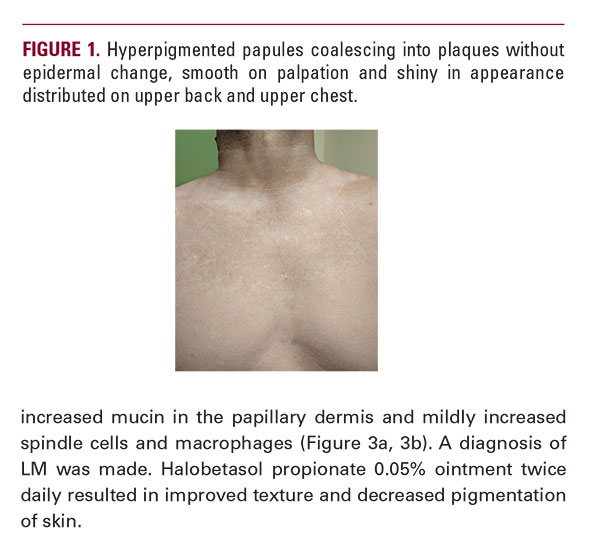
We present the case of a 66-year-old Hispanic male referred by his primary care physician for evaluation of asymptomatic dark spots on his trunk and extremities present for about one-year. Physical exam revealed smooth, brown hyperpigmented papules coalescing into plaques on the trunk. Multiple well-demarcated oval dark brown plaques measuring 3 cm in size were located on the upper back, peri-umbilical area, bilateral lower extremities, and buttocks. A diagnosis of lichen myxedematosus was made based on histologic features observed in the dermis.
There are 5 subtypes of LM: a discrete papular form, acral persistent papular mucinosis, self-healing papular mucinosis, papular mucinosis of infancy, and a pure nodular form. Occasional patients with LM have atypical features or features intermediate between scleromyxedema and localized LM. We present a case of atypical LM with mixed features of the different subtypes. Herein we will review the varied clinical presentations of LM and highlight the distinguishing features of scleromyxedema.
J Drugs Dermatol. 2020;19(3): 320-322 doi:10.36849/JDD.2020.4864
INTRODUCTION
Lichen Myxedematosus (LM), also referred to as papular mucinosis, was first described in 1906 by Dubreuilh. In 1953, Montgomery and Underwood classified LM into subsets which were revised in 2001 by Rongioletti and Rebora. Rongioletti and Rebora categorized LM into three subsets: 1) scleromyxedema, 2) localized, and 3) atypical.1 LM is characterized by lichenoid papules, nodules, and/or plaques due to dermal mucin deposition with varying degrees of fibrosis in the absence of thyroid disease.2 We present the case of a patient diagnosed with LM.
CASE REPORT
A 66-year-old Hispanic male presents with a one-year history of asymptomatic dark spots on his trunk and extremities. Physical exam revealed smooth papules coalescing into thin plaques on the trunk (Figure 1). Multiple well-demarcated oval dark brown plaques measuring about 2-3 cm in size were located on the upper back, periumbilical area, bilateral lower extremities and buttocks (Figure 2a, 2b). Lab values were within normal limits and showed a normal electrophoretic pattern. Histology showed increased mucin in the papillary dermis and mildly increased spindle cells and macrophages (Figure 3a, 3b). A diagnosis of LM was made. Halobetasol propionate 0.05% ointment twice daily resulted in improved texture and decreased pigmentation of skin.